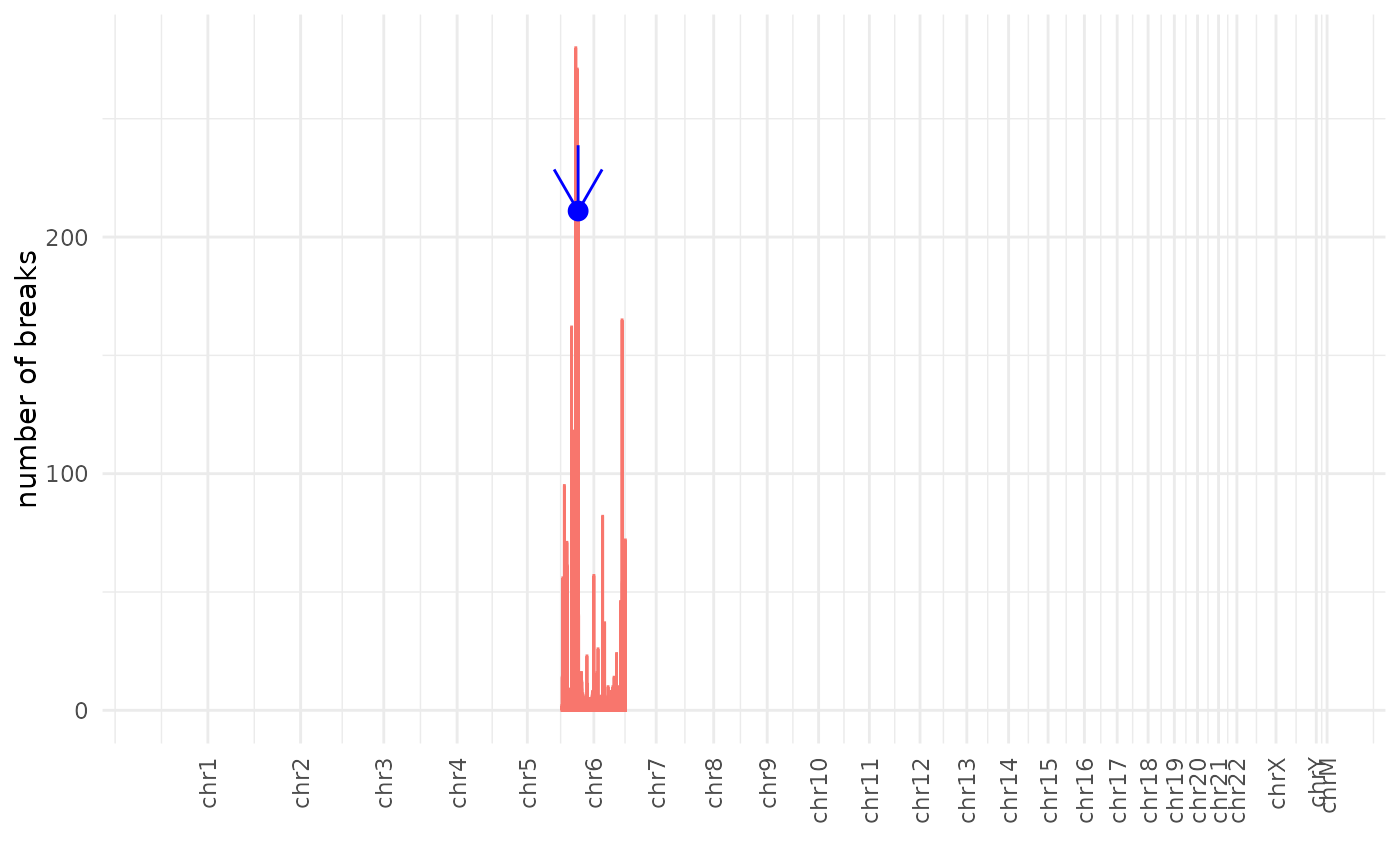
manhattan_plot Manhattan plot showing on/off-targets detected organized by chromosomal position with bar height representing read count (number of breaks).
manhattan_plot.Rdmanhattan_plot Manhattan plot showing on/off-targets detected organized by chromosomal position with bar height representing read count (number of breaks).
Usage
manhattan_plot(
x,
bsgenome,
standard_chromosomes = TRUE,
cutsite_breaks_only = TRUE,
subtract_nontarget = FALSE,
log_signal = FALSE,
verbose = TRUE
)Arguments
- x
A GRanges object containing the results of a breakinspectoR analysis. Possibly filtered (eg. qval < .01).
- bsgenome
character, bsgenome to use (eg. BSgenome.Hsapiens.UCSC.hg38).
- standard_chromosomes
logical, constrain the analysis to standard. chromosomes only. Default: TRUE.
- cutsite_breaks_only
display breaks of cutsite only, instead of using breaks accumulated in the complete protospacer. Default: TRUE
- subtract_nontarget
subtract breaks of non-target library. Default: FALSE
- log_signal
display log2 transformed signal. Default: FALSE.
- verbose
logical, keep informing about every step.
Examples
if (FALSE) { # \dontrun{
offtargets <- breakinspectoR(
target =system.file("extdata/vegfa.chr6.bed.gz", package="breakinspectoR"),
nontarget=system.file("extdata/nontarget.chr6.bed.gz", package="breakinspectoR"),
guide ="GACCCCCTCCACCCCGCCTC",
PAM =c("NGG", "NAG"),
bsgenome ="BSgenome.Hsapiens.UCSC.hg38",
cutsiteFromPAM=3
)
} # }
data(breakinspectoR_examples)
manhattan_plot(
offtargets,
bsgenome ="BSgenome.Hsapiens.UCSC.hg38"
)
#> Loading BSgenome.Hsapiens.UCSC.hg38...
#> done